Follow-up results of patients diagnosed with multicystic dysplastickidney disease
Prognosis in patients with multicystic dysplastic kidney disease
Authors
Abstract
AimMulticystic dysplastic kidney disease MCDKD is the second most common urinary system anomaly detected by prenatal USG and is probably one of the leading causes of having a single kidney in adults. When congenital kidney and urinary system anomalies are diagnosed in the intrauterine period and followed up and treated early in the postnatal period, the morbidity and mortality rates of the patients decrease and their quality of life increases. In this study, we aimed to evaluate the follow-up results of patients who were followed up with the diagnosis of MCDKD with prenatal and postnatal diagnosis, and to determine possible complications and their relationship with the prognosis.
MethodsIn this study, 31 patients with MCDKD were examined. Nineteen patients were male (61.2%), 25 (77.41%) patients were diagnosed in the prenatal period. The cases, which were diagnosed in the prenatal or postnatal period but were included in the study according to the hospital file and outpatient card information, were evaluated retrospectively.
ResultsMCDKD was detected on the left side in 18 patients (58.05%) and bilateral in one patient (3.2%). The follow-up period ranged from 7 days to 9 years. When the contralateral kidneys of the patients were examined, stage II hydronephrosis was found in two (6.6%), compensatory hypertrophy in 24 (80%) and the contralateral kidneys in 6 (20%) patients were found to be normal. It was found that hypertension developed in three (9.6%) patients and 15 (48%) patients had recurrent urinary tract infections during follow-up. Renal failure was detected in only one patient with bilateral MCDKD, and the patient died.
ConclusionIn conclusion, patients with MCDKD should be monitored with blood pressure, kidney function tests, urine analysis and USG at regular intervals. Early detection of complications such as hypertension, UTI, kidney failure, malignancy and appropriate treatment are important for the prognosis of the patients.
Keywords
Introduction
When congenital kidney and urinary system anomalies are diagnosed in the intrauterine period and followed up and treated early in the postnatal period, the morbidity and mortality rates of the patients decrease and their quality of life increases. Early diagnosis is difficult due to the asymptomatic course of many congenital anomalies of the kidney and urinary system. Today, with the introduction of advanced diagnostic methods, it has become possible to detect congenital anomalies before birth. The MKDKD incidence is approximately 1/ 2000. It is generally unilateral.1 Left kidney involvement is more frequent and is more common in males.2,3 Kidney function is provided by the contralateral, often compensatory, hypertrophied kidney. Bilaterals are incompatible with life. The cause of MKDKD is unclear. There may be an underlying genetic predisposition.4,5 It is a non-genetic condition that occurs sporadically.1 The incidence of genitourinary system anomalies has been reported to be high in patients with multicystic dysplastic kidneys.6,7,8 The syndromes can be seen together with urinary system malformations and non-urinary system malformations. The most common anomaly among these is vesicourethral reflux (VUR) with a rate of 18-43%.9,10 Generally, most of them are detected incidentally during prenatal ultrasonography (USG). The diagnosis is made by USG. Renal scintigraphy and voiding cystourethrography (VCUG) are routinely recommended to determine the renal function and the presence of VUR.11 Complications of multicystic dysplastic kidney disease are hypertension (HT), malignancy, kidney failure, and urinary tract infections (UTI).1 After diagnosis, patients should undergo renal USG at regular intervals to follow up urinalysis, kidney functions, blood pressure measurement and the development of hydronephrosis, tumor, compensatory hypertrophy in the contralateral kidney. In this study, it was aimed to evaluate the follow-up results of patients who were followed up with the diagnosis of MCDKD with prenatal and postnatal diagnosis, and to determine possible complications and their relationship with the prognosis. Early detection of complications such as hypertension, UTI, kidney failure, malignancy and appropriate treatment are important for the prognosis of the patients.
Materials and Methods
In our study, we examined 31 patients with multicystic dysplastic kidneys aged between 1 day and 14 years, who were diagnosed and followed up at any stage of the prenatal period or childhood for nine years at the Pediatric Nephrology Outpatient Clinic. The cases, which were diagnosed in the prenatal or postnatal period, but were included in the study according to the hospital file and outpatient card information, were evaluated retrospectively. No limit was determined for the follow-up period of the patients included in the study. Patients’ gender, age, age of diagnosis, duration of follow-up, week of gestation when prenatal MCDKD was detected, if diagnosed in the postnatal period, the reason for admission, accompanying anomaly, consanguinity of parents, family history, nephrectomy, number of UTIs, UTI prophylaxis and duration, hypertension, and kidney impairment were evaluated in terms of failure, tumor and family kidney disease. Urinalysis, kidney function tests, which side had the MCDKD, an anomaly in the opposite kidney, VCUG, MAG-3 and DMSA findings were investigated. USG was performed to show the presence of many unrelated cysts of different sizes in the normal anatomical location, rarely in the ectopic location, and without normal parenchyma. In the advanced period, it was performed in the atrophic kidneys. After catheterization of the bladder for VCUG, it was filled with a contrast agent diluted with saline from a height of 80 cm with free flow. Anteroposterior, right-left radiographs were obtained under fluoroscopy during voiding. DMSA and MAG-3 renal scintigraphy was performed to show the nonfunctioning kidney on the side of the disease. The Toshiba Xario brand USG device was used in the ultrasonographic examinations performed in our hospital in the postnatal period. DMSA and MAG-3 renal scintigraphy was performed with Siemens Optilux and GE brand devices.
Results
In our study, 31 patients with multicystic dysplastic kidneys aged 1 day to 14 years (mean age 5.4) who were incidentally diagnosed and monitored between 2000-2009 were included. Nineteen patients were male (61.2%), 12 were female (38.7%), and the male/female ratio was 1.58. Twenty-five (77.41%) patients were diagnosed in the prenatal period and 6 (22.52%) in the postnatal period. It was determined that one of the patients diagnosed in the postnatal period was diagnosed during UTI and 5 during abdominal USG performed for various reasons. The age of diagnosis in 6 patients diagnosed in the postnatal period ranged from 3 months to 11 years. MCDKD was detected on the left side in 18 patients (58.05%), on the right side in 12 patients (38.74%), and bilateral in one patient (3.2%). The follow-up period ranged from 7 days to 9 years (mean 4.8 years) (Table-1). When the contralateral kidneys of the patients were examined by USG, 2 (6.6%) were detected to have stage II hydronephrosis, 24 (80%) compensatory hypertrophy and 6 (20%) had normal contralateral kidneys. It was observed that hydronephrosis spontaneously recovered during follow-up (Table 2).VCUG results of 20 patients were obtained, and it was learned that none of them had VUR. VCUG was planned for the remaining 11 patients but could not be performed because consent from the families could not be acquired. MAG-3 scintigraphy was performed in 20 patients, and dysfunctional kidneys were detected on the side with MCDKD in all patients. In DMSA applied to five patients, the kidney on the side with MCDKD could not be scanned. In the IVP performed on one patient, no function was detected on the side with MCDKD. During the follow-up, hypertension developed in three (%9,6) patients. When there was no response to antihypertensive treatment, nephrectomy was performed. The blood pressure of the patients returned to normal after nephrectomy. Fifteen patients were found to have urinary tract infection during follow-up. It was determined that the infection recurred on average 1.6 times a year, and the patients received antibiotic prophylaxis for 1-2 years. Kidney function tests in 30 patients were normal. Only 1 patient with bilateral MCDKD had renal failure and this patient died on the 7th day of his life. During the followup period, no malignancy developed in any of the patients (Table 3). There was a second-degree consanguinity between the parents of two patients, and a third-degree consanguinity in two patients. Parents and siblings of 19 patients were screened for MCDKD by USG. MKDBH was not detected in any patient. Polycystic kidney disease was detected in one patient’s mother, sibling and maternal uncle. The grandfather of two patients and the cousin of one patient had a history of CRF, and the etiology was not available. It was found that MCDKD accompanied asthma and obesity in one patient, growth retardation in two, mental motor retardation and VSD in one, PDA, ASD and cryptorchidism in one, and ASD, VSD and hydrocephalus in one.
Discussion
When patients with congenital kidney and urinary system anomalies are diagnosed in the intrauterine period and followed up and treated early in the postnatal period, their morbidity and mortality rates decrease and their quality of life increases. In connection with the increase of pregnancy follow-up in recent years, congenital anomalies of kidney and urinary system are more frequently diagnosed in the intrauterine period. MCDKD is one of them. MCDKD is the second most common urinary system anomaly detected by prenatal USG, and is probably one of the leading causes for the presence of a single kidney in adults.11 Patients are more often diagnosed prenatally, and less frequently incidentally by USG, which is performed for various reasons. In the study conducted by Onal et al., it was reported that 80% of 61 patients who were followed up with MCDKD, were diagnosed in the prenatal period and 20% in the postnatal period.10,12 In our study, 77.4% of our patients were diagnosed in the prenatal period, and 22.52% in the postnatal period with UTIs and USG performed for various reasons.
The incidence of MCDKD is 1/2000, and it is usually unilateral.1 Left side involvement is more frequent and more common in males. The male/female ratio is 1.48.13 In our study, 61.2% of our patients were male, 38.7% were female, and the male/female ratio was 1.58. MCDKD was left-sided in 58% of the patients. In a study by Ismaili et al., 58% of the patients were male and 46% of the patients had right-sided MCDKD.14 In a study by Miller et al., 59% of the patients were male, 64% of the patients had right-sided MCDKD.15 and in the study by Onal et al., 70% of the patients were male and 54% of the patients were right-sided.12 While it is more common in the male gender, it is seen that the location varies.in different studies. Urinary malformations accompanied by MCDKD include VUR, ureteropelvic stenosis, renal agenesis, renal hypoplasia, renal dysplasia, horseshoe kidney, ureterocele, ectopic kidney, bladder diverticulum, urethral valve, fibromuscular dysplasia, and seminal vesicle abnormalities.13 The most common anomaly among these is VUR with a rate of 18-43%. In patients with unilateral MCDKD, it is recommended to examine the contralateral kidneys in terms of VUR, UPJ and UVB stenosis, size and function of the contralateral kidney.10 It was observed that the hydronephrosis, detected in 2 of our patients, regressed spontaneously in the follow-up, and VUR was not detected in both patients. There was no urinary system anomaly in addition to MCDKD in these patients. Hypertension is an important complication of MCDKD. Studies conducted in recent years show that the risk of HT in MCDKD is lower than what is known.12,14,15,16 In a meta-analysis evaluating 29 studies, the rate of hypertension in MCDKD was reported as 5.4 per 1000.16 Although there are publications reporting that hypertension can be treated with nephrectomy, there are also publications showing that hypertension may continue despite nephrectomy. Snodgrass performed nephrectomy in 4 patients with MCDKD and hypertension and reported that 2 patients had improved hypertension and the other 2 patients still had elevated blood pressure.17 In our study, hypertension was detected in 3 (10%) of our 30 unilateral MCDKD patients and nephrectomy was performed in all three of them because they did not benefit from antihypertensive treatment. Blood pressure returned to normal after nephrectomy. In our patient group, hypertension was found at a higher rate than expected. Even though it is rare, there still is a risk of malignancy in MCDKD. These are mostly Wilms tumors and renal cell carcinoma.11 Cystic kidney diseases such as acquired cystic kidney disease, tuberous sclerosis and von Hippel-Lindau are highly preneoplastic, and the rate of kidney tumor development is high in these cases. Neoplasia was not encountered in our study, either. Due to the low risk of tumor development, nephrectomy is not recommended in MCDKD, and it is thought that follow-up with USG is sufficient. Another important complication of MCDKD is UTI. The frequency of UTI is reported to be 3.6-19%.18 MCDKD can be diagnosed incidentally with USG performed during UTI. Urinary system infection was detected in 48% of our patients. Bilateral MCDKD is very rare but incompatible with life. Infants are usually stillborn or lost in the first few hours of life.1 Our patient with bilateral MCDKD lived for 7 days. There is no definite data on the necessity of nephrectomy in MCDKD. In addition to complications such as hypertension and malignancy, there are authors who recommend nephrectomy to reduce longterm follow-up costs. However, due to the rarity of complications, it has been reported that follow-up with close blood pressure and USG is sufficient. The American Academy of Pediatric Urology also recommends nephrectomy for cases with MCDKD in cases such as abdominal pain, UTI, hypertension, and renal malignancy.16 MCDKD can be seen together with syndromes (VACTERL, Branchio-Auto-Renal (BOR) syndrome, Williams syndrome, Beckwith-Wiedemann syndrome, Trisomy 18.49 syndrome).13 No syndromes were found in our patients. Malformations outside of the urinary system may also accompany MCDKD. These include the gastrointestinal system (esophageal atresia tracheaesophageal fistula, duodenatresia, imperforeanus, omphalocele, inguinal hernia and Hirschsprung’s disease), cardiovascular system (VSD, PDA, AS, PS and aortic coarctation), neurological system (anencephaly, microcephaly, microcephaly) anomalies of the musculoskeletal system (syndactyly, congenital hip dislocation and pes equinovarus) and micrognathia, cleft palate, hymenalatresia, and uterine anomalies.13 In our study, it was found that MCDKD accompanied asthma and obesity in one patient, growth retardation in two, mental motor retardation and VSD in one, PDA, ASD and cryptorchidism in one, and ASD, VSD and hydrocephalus in one. MCDKD undergoes involution over time, and the contralateral, often compensatory, hypertrophied kidney takes over the functions of both kidneys. Compensatory hypertrophy has been reported with a rate of 43-100%.19,20 Compensatory hypertrophy was found in 80% of our patients. It is a sporadic and non-genetic condition.1 However, familial cases have also been reported. It is considered that it may be based on genetic attachment.4,5 There are publications indicating different members of a family having MCDKD. This information may support the role of genetic factors in the formation of familial MCDKD. In a study by Sekine et al., it was reported that the father of a patient with unilateral MCDKD also had unilateral MCDKD, the mother had a previous miscarriage, and bilateral MCDKD was detected in the autopsy of the fetus. The presence of MCDKD in 3 people in a family suggests the presence of hereditary factors.21 MCDKD was not found in the family history of our patients and in renal USGs performed on parents and siblings. Autosomal dominant polycystic kidney disease was found in the mother, maternal uncle and brother of one of our patients. In conclusion, the widespread use of antenatal USG enables early diagnosis and treatment of urinary system pathologies and reduces mortality-morbidity rates. Whether diagnosed incidentally in the prenatal or postnatal period, patients with MCDKD should be monitored with blood pressure, kidney function tests, urine analysis and USG at regular intervals. Early detection of complications such as hypertension, UTI, renal failure, malignancy and appropriate treatment are important for the prognosis of the patients.
Declarations
Animal and Human Rights Statement
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. No animal or human studies were carried out by the authors for this article.
Data Availability
The datasets used and/or analyzed during the current study are not publicly available due to patient privacy reasons but are available from the corresponding author on reasonable request.
Conflict of Interest
None of the authors received any type of financial support that could be considered potential conflict of interest regarding the manuscript or its submission.
Funding
None.
Scientific Responsibility Statement
The authors declare that they are responsible for the article’s scientific content including study design, data collection, analysis and interpretation, writing, some of the main line, or all of the preparation and scientific review of the contents and approval of the final version of the article.
References
-
Elder JS. Congenital anomalies and dysgenesis of the kidney. In: Kliegman RM, Stanton BF, St Geme JW III, Schor NF, editors. Nelson textbook of pediatrics. 20th ed. Philadelphia, PA: Elsevier; 2016:2554-2570.
-
Schreuder MF. Unilateral anomalies of kidney development: why is left not right? Kidney Int. 2011;80(7):740-745. doi:10.1038/ki.2011.204
-
Van Eijk L, Cohen-Overbeek TE, Hollander NS, Nijman JM, Wladimiroff JW. Unilateral multicystic dysplastic kidney: a combined pre- and postnatal assessment. Ultrasound Obstet Gynecol. 2002;19(2):180-183. doi:10.1046/j.0960-7692.2001.00497.x
-
Hwang DY, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 2014;85(6):1429-1433. doi:10.1038/ki.2013.508
-
Xi Q, Zhu X, Wang Y, Ru T, Dai C, Wang Z, et al. Copy number variations in multicystic dysplastic kidney: update for prenatal diagnosis and genetic counseling. Prenat Diagn. 2016;36(5):463-468. doi:10.1002/pd.4807
-
Ranke A, Schmitt M, Didier F, Droulle P. Antenatal diagnosis of multicystic renal dysplasia. Eur J Pediatr Surg. 2001;11(4):246-254. doi:10.1055/s-2001-17147
-
Shibata S, Shu Y, Shigeta M, Watanabe T, Nagata M. Initial pathological events of renal dysplasia with urinary tract obstruction in utero. Virchows Arch. 2001;439:560-570. doi:10.1007/s004280100420
-
Mathiot A, Liard A, Eurin D, Docher JN. Prenatally detected multicystic renal dysplasia and associated anomalies of the genitourinary tract. J Radiol. 2002;83:731-735.
-
Kara A, Gurgoze MK, Aydin M, Koc ZP. Clinical features of children with multicystic dysplastic kidney. Pediatr Int. 2018;60(8):750-754. doi:10.1111/ped.13612
-
Atiyeh B, Husmann D, Baum M. Contralateral renal abnormalities in multicystic dysplastic kidney. J Pediatr. 1992;121(1):65-067. doi:10.1016/s0022-3476(05)82543-0
-
Fryer K, Nield LS, Muchant DG. Multicystic dysplastic kidney. Clin Pediatr (Phila). 2007;46(4):365-367. doi:10.1177/0009922806290101
-
Onal B, Kogan BA. Natural history of patients with multicystic dysplastic kidney: what follow-up is needed? J Urol. 2006;176(4 Pt 1):1607-1611. doi:10.1016/j.juro.2006.06.035
-
Robson WL, Leung AK, Thomason MA. Multicystic dysplasia of the kidney. Clin Pediatr (Phila). 1995;34(1):32-034. doi:10.1177/000992289503400106
-
Ismaili K, Avni FE, Alexander M, Schulman C, Collier F, Hall M. Routine voiding cystourethrography is of no value in neonates with unilateral multicystic dysplastic kidney. J Pediatr. 2005;146(6):723-725.
-
Miller DC, Rumohr JA, Dunn RL, Bloom DA, Park JM. What is the fate of the refluxing contralateral kidney in children with multicystic dysplastic kidney? J Urol. 2004;172(4 Pt 2):1630-1634. doi:10.1097/01.ju.0000138818.10910.1f
-
Narchi H. Risk of hypertension with multicystic kidney disease: a systematic review. Arch Dis Child. 2005;90(9):921-924. doi:10.1136/adc.2005.075333
-
Snodgrass WT. Hypertension associated with multicystic dysplastic kidney in children. J Urol. 2000;164(2):472-473. doi:10.1016/s0022-5347(05)67402-2
-
Tilemis S, Savanelli A, Baltogiannis D, Cigliano B, Settimi A. Is the risk of hypertension an indication for prophylactic nephrectomy in patients with unilateral multicystic dysplastic kidney? Scand J Urol Nephrol. 2003;37(5):429-432. doi:10.1080/00365590310006282
-
Kiyak A, Yilmaz A, Turhan P, Sander S, Aydin G, Aydogan G. Unilateral multicystic dysplastic kidney: single-center experience. Pediatr Nephrol. 2009;24(1):99-104. doi:10.1007/s00467-008-0942-7
-
Wacksman J, Phipps L. Report of the multicystic kidney registry: preliminary findings. J Urol. 1993;150(6):1870-1872. doi:10.1016/s0022-5347(17)35918-9
-
Sekine T, Namai Y, Yanagisawa A, Shirahama H, Tashiro Y, Terahara M, et al. A familial case of multicystic dysplastic kidney. Pediatr Nephrol. 2005;20(9):1245-1248. doi:10.1007/s00467-005-1905-x
Tables
Table 1. Demographic Characteristics of Patients

n: Total number of patients
Table 2. Urological anomalies accompanying MCDKD
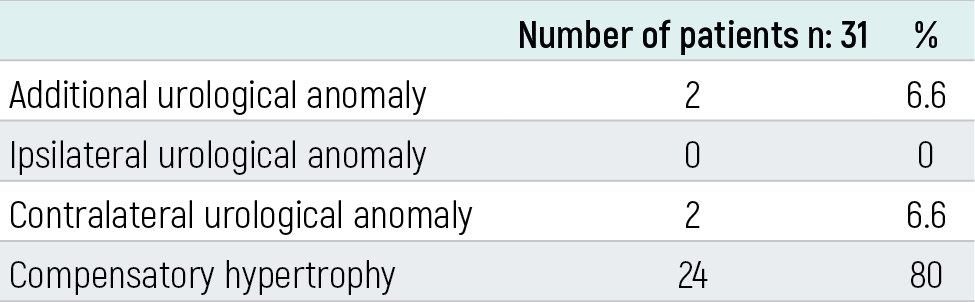
n: Total number of patients
Table 3. Complications in MCDKD patients
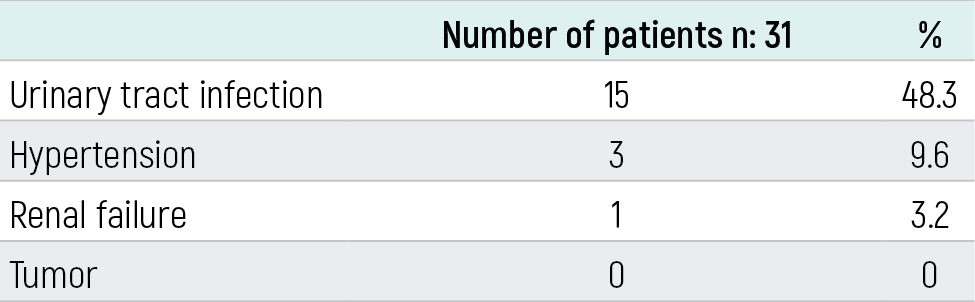
n: Total number of patients
Additional Information
Publisher’s Note
Bayrakol MP remains neutral with regard to jurisdictional and institutional claims.
Rights and Permissions

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License (CC BY-NC 4.0). To view a copy of the license, visit https://creativecommons.org/licenses/by-nc/4.0/
About This Article
How to Cite This Article
Ümmühan Çay, Nilgün Çakar. Follow-up results of patients diagnosed with multicystic dysplastickidney disease. Eu Clin Anal Med 2021;9(2):11-13. doi:10.4328/ECAM.10028
- Received:
- March 1, 2021
- Accepted:
- March 18, 2021
- Published Online:
- April 16, 2021
- Printed:
- May 1, 2021